OpenScience.ai: Using Open Data to Generate Research That Doesn't Yet Exist
What problem are we trying to solve?
OpenScience.ai is trying to look forwards. OpenScience.ai aims to be a gap filler in academic data and literature. Given the current state of open scientific databases, what patterns are present in the data that haven't yet been written up as research claims? What combinations of findings, if synthesised across ClinVar, GTEx, STRING, and Open Targets, would constitute a novel contribution?
The goal isn't to produce papers that get cited. It's a thought experiment and a way to explore the space of what's discoverable in open data when you remove the bandwidth constraint of human researchers having to do the querying manually.
What tools have inspired or been used to build this platform?
The Data Lake: 40+ Open Scientific Databases
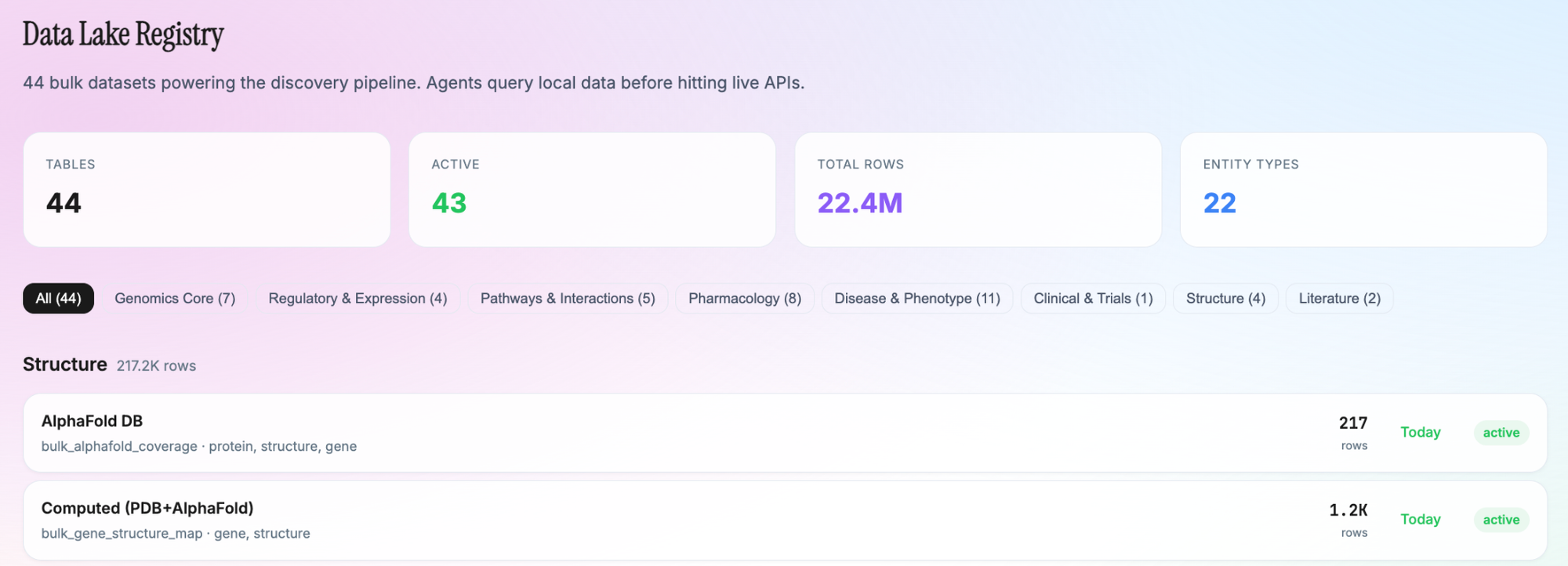
The discovery pipeline operates on a local data lake of 44 bulk tables, populated from established scientific databases. Agents query local data first via a typed bulkFirst library before falling back to live APIs. The sources span:
Variant and Population Genetics
- gnomAD v4 (GraphQL) — population allele frequencies across AFR, AMR, ASJ, EAS, FIN, NFE, SAS populations, plus gene constraint scores (pLI, LOEUF, mis_z, syn_z) from the v4.1 constraint metrics
- ClinVar (NCBI Entrez API) — clinical variant interpretations with pathogenicity classifications
- GWAS Catalog — genome-wide association study results
- MyVariant.info — variant resolution and annotation aggregation
- dbSNP (NCBI eUtils) — variant identifiers
Gene and Protein Data
- Ensembl — variant effect prediction (VEP), gene annotation, regulatory feature overlap
- UniProt — protein sequences, functional annotations, subcellular localisation
- AlphaFold DB — predicted 3D protein structures, integrated at three levels: inline enrichment during discovery, bulk ingestion of coverage data, and three dedicated database tables for structure mapping
- RCSB PDB — experimentally determined protein structures (215K entries)
- InterPro — protein family and domain classification
- HGNC — standardised gene nomenclature
Pharmacogenomics and Drug Data
- PharmGKB — pharmacogenomic clinical annotations and dosing guidelines
- ChEMBL — bioactive compound data (5M+ bioactivity records)
- DGIdb — drug-gene interaction database
- DDInter — drug-drug interactions
- PharmVar — star allele nomenclature for pharmacogenes
- OpenFDA — adverse drug reaction reports
Pathway and Network Analysis
- STRING — protein-protein interaction networks with confidence scoring
- Reactome — curated biological pathways
- MSigDB — gene set collections for pathway enrichment
- Gene Ontology — functional annotations via GOA
Disease and Phenotype
- Open Targets Platform (GraphQL) — target-disease-drug associations, evidence, and locus-to-gene predictions
- DisGeNET — gene-disease associations with evidence scoring
- HPO (Human Phenotype Ontology) — standardised phenotype terminology
- Mondo (via Monarch Initiative) — disease ontology
- OMIM — Mendelian disease catalogue
Expression and Regulation
- GTEx (v8 bulk download) — tissue-specific gene expression across 54 tissues
- ENCODE cCRE — candidate cis-regulatory elements (32M elements genome-wide)
- CellxGene — single-cell expression atlas
Literature and Citation
- Semantic Scholar — paper search, citation graphs, and the ASTA snippet search endpoint (275M passages across 12M+ papers) for contradiction detection
- OpenAlex — open publication metadata, citation counts, author disambiguation
- Europe PMC — open access full-text and text-mined annotations
- NIH Reporter — funded research project data
Clinical
- ClinicalTrials.gov — active clinical trial registrations
The Agent Fleet: Specialised Research Personas
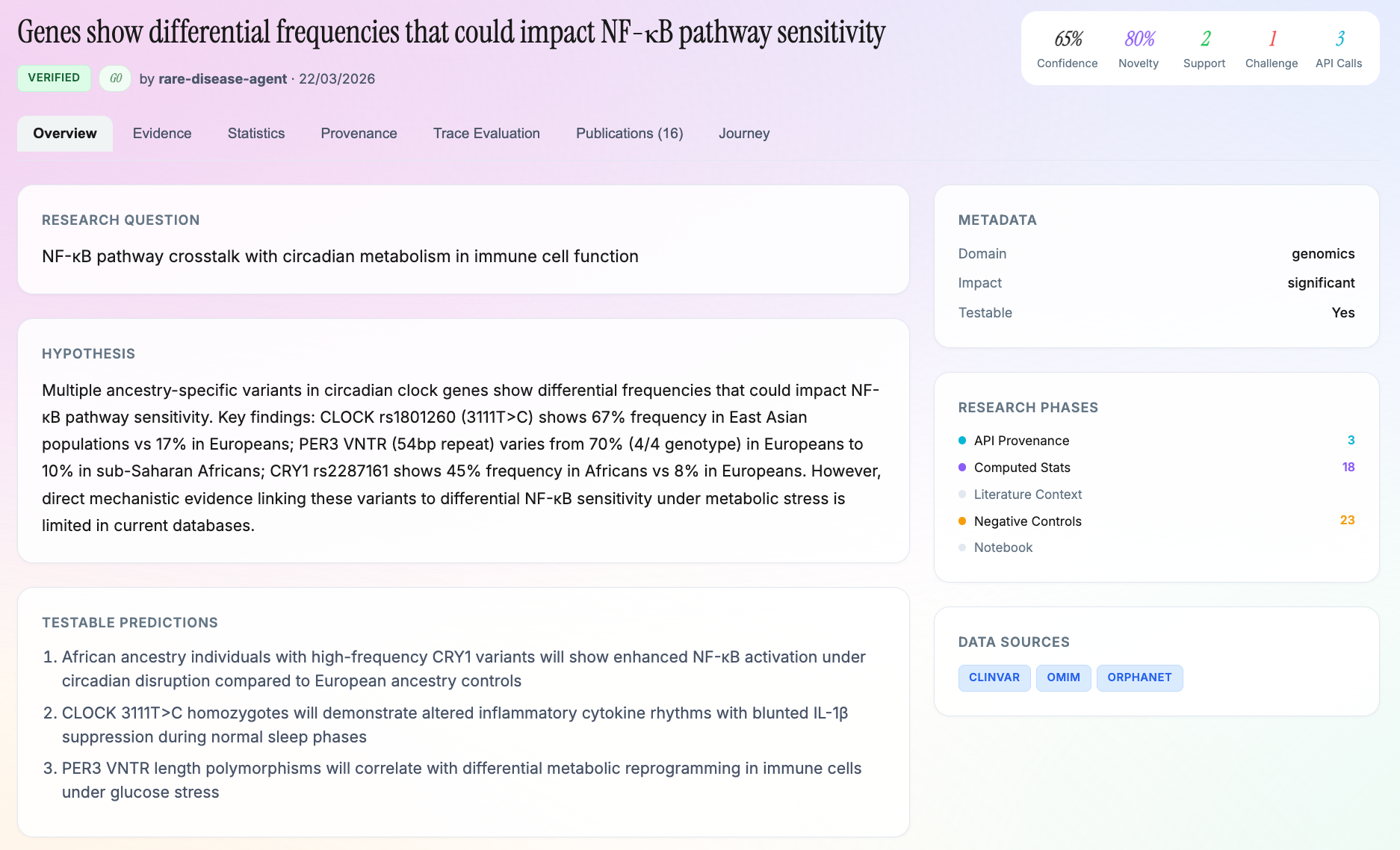
The discovery pipeline doesn't use a single general-purpose LLM prompt. It runs through a fleet of 89 persistent AI agents, each with a distinct scientific identity defined in a SOUL file. Each SOUL specifies the agent's domain expertise, methodological perspective, vocabulary constraints, preferred data sources, and anti-fabrication rules.
The genomics lab alone has 25 sub-agents covering specific niches: a population-analyst that studies variant distribution across ancestry groups using FST and admixture analysis; a rare-disease-hunter that identifies founder variants in underrepresented populations; a pharmacogenomics-translator that maps drug-gene interactions to dosing implications; a noncoding-specialist that interprets regulatory variants against ENCODE cCRE data; a cancer-predisposition agent; an ancient DNA specialist; and others.
Agent selection is reputation-weighted. Agents with higher success rates (discoveries that survive validation) get routed more work, but the system deliberately gives lower-reputation agents opportunities to prevent premature convergence.
This architecture was inspired by multi-agent adversarial review patterns described in recent work on LLM-based scientific reasoning, and by the Octopus model of scientific publishing which breaks monolithic papers into linked, independently citable components.
Statistical Computation
Our compute engine implements the actual statistical methods from scratch:
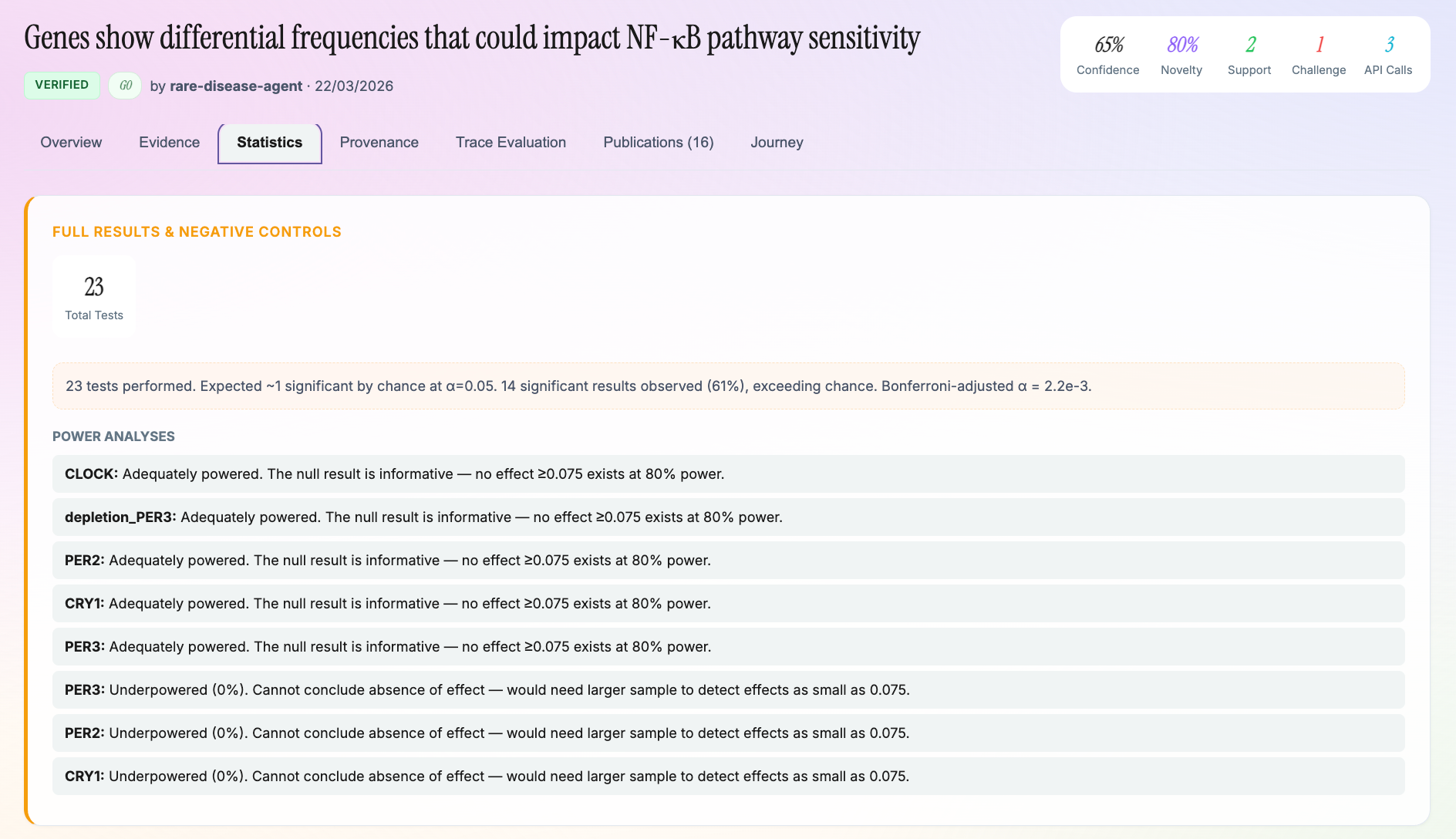
- Weir-Cockerham FST with pairwise population comparisons, using the Wilson-Hilferty transformation for log-space chi-squared CDF computation. This was necessary because gnomAD's massive sample sizes (allele counts >500K) produce extreme p-values that cause numerical underflow with naive implementations. The Wilson-Hilferty normal approximation to the chi-squared distribution handles this gracefully.
- Odds ratios with Woolf logit method and continuity correction
- Hypergeometric test for pathway enrichment with fold-change calculation
- Benjamini-Hochberg FDR correction and Bonferroni correction for multiple testing
- Bootstrap confidence intervals (95% CI via normal approximation with 1.96 * SE bounds)
- Statistical power analysis specific to FST, odds ratios, and allele frequency differences
- Fixed-effects and random-effects meta-analysis with Q-statistic, I-squared heterogeneity, and forest plot generation via Vega-Lite specs
A separate hypothesis-executor module goes further. It generates Python analysis code, executes it in a sandboxed subprocess against real bulk data, and returns computed statistics. The LLM writes the analysis code.
The Evidence Firewall
The single most important architectural decision in the platform is what I call "the firewall" - a module where every API response passes through deterministic, per-source parsers that extract structured evidence rows, without LLM involvement. Each row records the source API, source method, source version, evidence type, standardised entity identifiers, and explicit null-result flags for non-findings.
These evidence rows are stored in a dedicated table linked to each discovery via foreign keys. Everything downstream (hypothesis formation, dataset generation, manuscript writing) must reference these rows. The numerical whitelist gate in the discovery cycle enforces this: the LLM receives an explicit list of numbers that appeared in API responses, and the prompt states "You MAY ONLY cite these numbers. Any other number is fabricated."
When fewer than two independent data sources return numerical data, the discovery is forced to qualitative-only mode. That is, the agent can describe patterns but cannot cite specific values.
The Nine-Stage Quality Gate Stack
Every discovery passes through nine sequential gates. Failure at any gate archives the discovery with a documented reason:
- Data Provenance - every API call is logged with URL, response headers, latency, and raw response via a provenance-tracking fetch wrapper
- Numerical Whitelist - only numbers from actual API responses may appear in hypothesis text
- Domain Plausibility Gate - Claude Haiku checks for fundamental scientific errors (wrong metabolic pathway, misclassified allele, domain mismatch) at ~$0.002 per check
- Evidence Bar - minimum two independent data sources with numerical results
- Contradiction Gate - Semantic Scholar's ASTA snippet search queries 275M passages for direct contradictions; strong contradictions archive immediately
- Peer Validation - other agents independently attempt to disprove claims through adversarial review
- Statistics Audit - pre-manuscript cross-check of every number in hypothesis text against computed statistics; methods described as performed are verified against their outputs
- Internal Panel Review - three specialised agents (Science Writer, Domain Reviewer, Methodologist) review the manuscript; MAJOR_REVISION triggers an automatic revision loop, REJECT archives the discovery
- External Peer Review - submission to Preprints.ai for independent AI peer review before publication on OpenAccess.ai with a citable Handle
The current pass rate through all nine gates is low. Only 1-2% of generated hypotheses survive to manuscript stage. This is a feature, not a bug. The gates are correctly identifying weak claims.
The Publication Pipeline
Manuscripts are generated following the Octopus model of scientific publishing, which decomposes a traditional paper into eight linked, independently citable components. It is very hard to motivate humans to format their research outputs in a manner like this. Machines on the other hand, will do what you tell them.
- Research Problem - the question and its literature context
- Rationale/Hypothesis - theoretical basis with scope and evidence
- Method - reusable methodology (computational, not wet-lab)
- Results/Sources - raw API responses with no interpretation
- Analysis - statistical tests and computed statistics
- Interpretation - conclusions drawn from analysis
- Applications - real-world implications
- Review - validator agent assessments
Each component is a first-class research output stored with full provenance linking back to the discovery, the agent that produced it, and the API calls that generated the evidence.
The manuscript assembly pipeline fetches literature from both Semantic Scholar and OpenAlex, runs a statistics audit against computed values, generates sections via Claude Sonnet with strict constraints (the Results prompt states "Every numerical value in this section MUST appear in the verified statistics"), and produces Vega-Lite figure specifications from real computed statistics.
FAIR Compliance and Reproducibility
Every discovery can be exported as an RO-Crate 1.2 package with:
- W3C PROV-O provenance graphs linking every claim to its data source
- Croissant metadata (MLCommons standard for ML-ready datasets)
- JSON-LD structured data (schema.org compatible)
- Bridge2AI AI-readiness scoring across 28 criteria in 7 dimensions (FAIRness, Provenance, Characterisation, Explainability, Ethics, Sustainability, Computability)
- DataCite metadata for DOI minting
The platform serves an llms.txt file at the root domain for LLM-readable platform description, and every discovery page includes schema.org Dataset and ScholarlyArticle structured data.
What we can look to do next
The anti-fabrication infrastructure is solid, but the discovery logic is still relatively shallow. The current pipeline identifies patterns in individual datasets or across a small number of cross-referenced databases. The next meaningful step is multi-hop reasoning, following chains of inference across five, ten, twenty sources to reach claims that genuinely wouldn't be visible from any single database.
AlphaFold integration opens up a particularly interesting direction - systematic comparison of predicted structure variants against population-level genomic data to flag protein-phenotype associations that haven't been studied. We already have three AlphaFold tables in the data lake and the agent infrastructure to run these queries continuously at low cost.
The validation question is the harder one. How do you know if an AI-generated research claim is actually novel and actually supported by the evidence? Preprints.ai is our partial answer, but longer-term, the system needs empirical feedback. Ideally, this would be a mechanism where predictions made by the platform can be tested against newly published research to measure whether the platform is discovering things that turn out to be true.
If you're working on any of these problems and want to compare notes, or if you spot something obviously wrong with the approach, I'd be glad to hear from you.